No products in the cart.

You're probably here because a peptide sequence is sitting in front of you, an experiment is queued up, and you need one number you can trust. That number is the peptide's molecular weight. If it's wrong, your stock concentration is wrong, your mass spec comparison is off, and any downstream interpretation gets shaky fast.
A good peptide calculator molecular weight workflow isn't just about typing in a sequence and copying the result. It's about knowing which mass you need, what chemistry the calculator assumes, and how that number carries into reconstitution, QC, and instrument readouts. In practice, that's where most mistakes happen.
Accurate peptide molecular weight is the number that ties your peptide's structure to what you do in the lab. You use it when you prepare a stock, when you compare a theoretical mass to an observed signal, and when you check whether supplier documentation matches what you ordered.
A small misunderstanding early on can create a string of avoidable errors. The concentration you think you made may not be the concentration in the tube. The peak you think confirms identity may be mismatched because you selected the wrong mass convention. The amount you order for a study may not match the chemistry you intended.
Practical rule: Never treat “molecular weight” as a single universal value until you've confirmed the peptide form being measured.
For a simple linear peptide, the calculation is straightforward. In real research, peptides often aren't simple. Terminal modifications, oxidation state, phosphorylation, and other chemistry can change the correct answer in ways that matter during formulation, QC, and ordering.
That's why experienced researchers don't just ask, “What's the molecular weight?” They ask:
If you build that habit, the calculator becomes more than a convenience. It becomes part of experimental control.
The first fork in the road is deciding which mass you need. Most peptide calculator molecular weight tools can show average molecular weight and monoisotopic molecular weight, but they are not interchangeable.
Average molecular weight uses the weighted average masses of naturally occurring stable isotopes for each element in the peptide. This is often the more intuitive number for bulk material handling, especially when you're thinking about macroscopic amounts of powder.
Monoisotopic molecular weight uses the mass of the most abundant stable isotope of each element. This is the value researchers usually care about for high-resolution mass spectrometry, because the instrument resolves isotope patterns and the exact peak assignment depends on the isotope-defined mass.
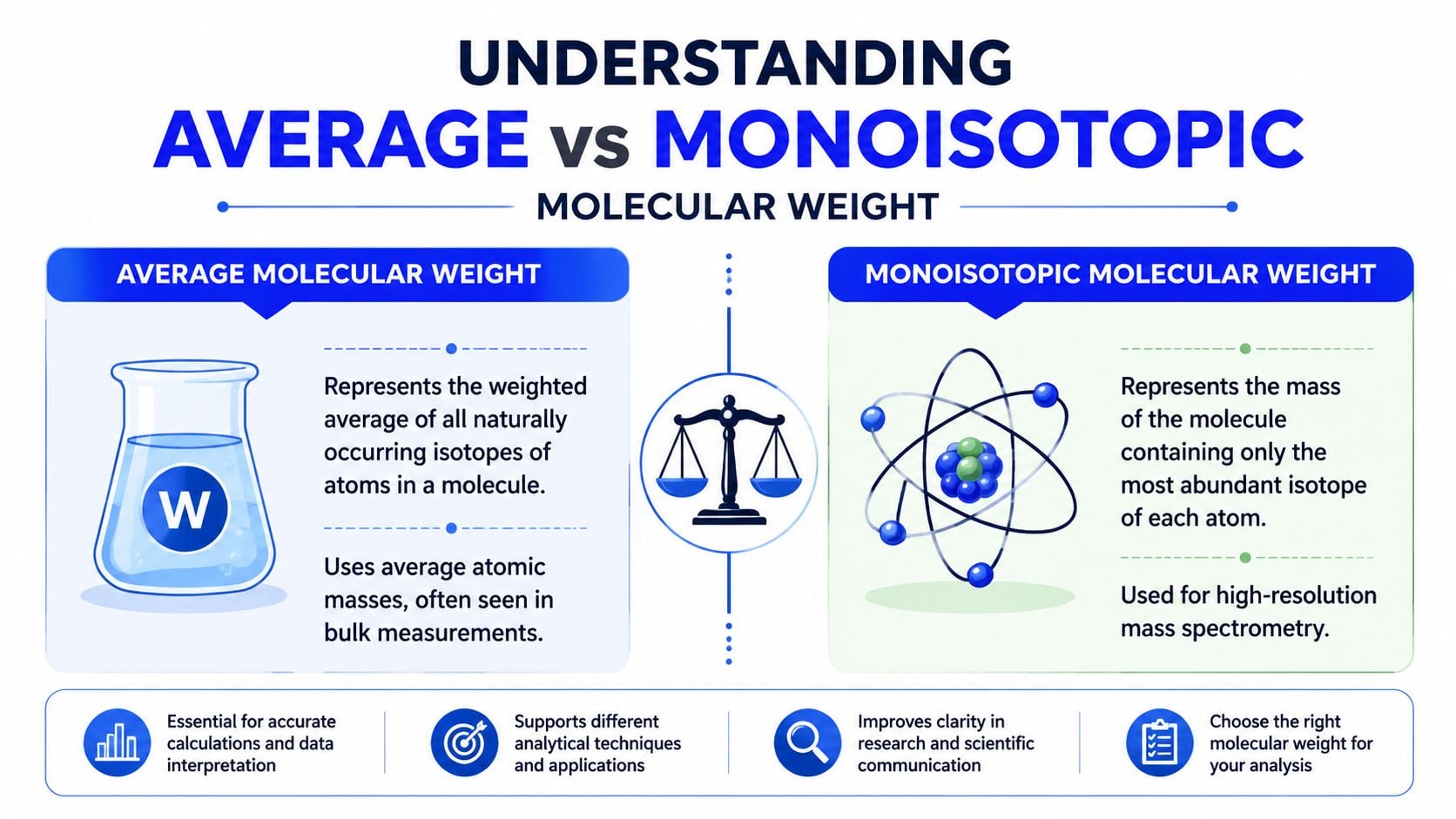
A useful way to think about it is this. Average mass reflects what a population of molecules looks like in aggregate. Monoisotopic mass reflects one specific isotope-defined version of that molecule. Neither is “more correct” in every context. The right one depends on the task.
| Mass type | Best used for | Why it matters |
|---|---|---|
| Average molecular weight | Bulk weighing, routine solution planning | Matches the way many researchers think about a lot of material as a whole |
| Monoisotopic molecular weight | High-resolution MS review, peak assignment | Aligns with isotope-specific interpretation of instrument data |
Use monoisotopic mass when you're checking a precise MS signal. Use average mass when you're discussing the peptide more generally for handling and preparation, unless your internal method says otherwise.
What doesn't work is switching between the two without noticing. That leads to confusing comparisons, especially when someone calculates one value and then tries to validate against data generated under the other convention.
A second problem comes from calculators that present a result without making the mass type obvious. Before you copy a number into a worksheet or protocol, check the label. “MW” by itself isn't enough.
Manual calculation is worth learning even if you rely on software every day. It teaches you what the calculator is doing, and it makes it much easier to catch errors when a result looks strange.
For a simple linear peptide, the basic approach is:
That last step matters because peptide bonds form by loss of water during chain assembly. When you calculate the mass of the intact final peptide, you work from residue masses and then add back the mass of one water molecule for the complete molecule.

A manual check is especially useful when you're dealing with a short sequence. You can often spot sequence-entry mistakes, terminal-state confusion, or a modification that got dropped from the request form.
Here's the thought process for a simple tripeptide:
Manual math stops being reliable when the peptide is no longer a plain linear chain with free termini. Many research peptides include chemistry that changes the mass and changes what the number means in practice. Bachem's peptide calculator guidance notes that many public calculators are built around simple, unmodified peptides, while research use often involves N-terminal modifications, oxidized cysteines, phosphorylated residues, or other custom chemistry.
That matters because those changes aren't cosmetic. They alter the actual molecular weight and can affect formulation, QC, and ordering decisions.
Common examples that require adjustment include:
If the peptide name includes “Ac-”, “amide”, “phospho-”, “oxidized”, or a stated disulfide pattern, don't trust a plain-sequence calculation.
Many early-career researchers make the same mistake. They calculate the peptide they think they have, not the peptide that was synthesized. Those aren't always the same molecule.
For that reason, manual calculation works best as a logic check. It helps you understand the structure. It does not replace a calculator that explicitly supports the chemistry in your construct.
Daily lab work moves too fast for repeated hand calculation. An online peptide calculator molecular weight tool is the better choice when you need consistent outputs, fewer arithmetic mistakes, and clearer handling of peptide variants.
Early in the process, use a calculator to reduce transcription and math errors. Later in the process, use it to sanity-check what's on the order form, in the COA, or in your data sheet.
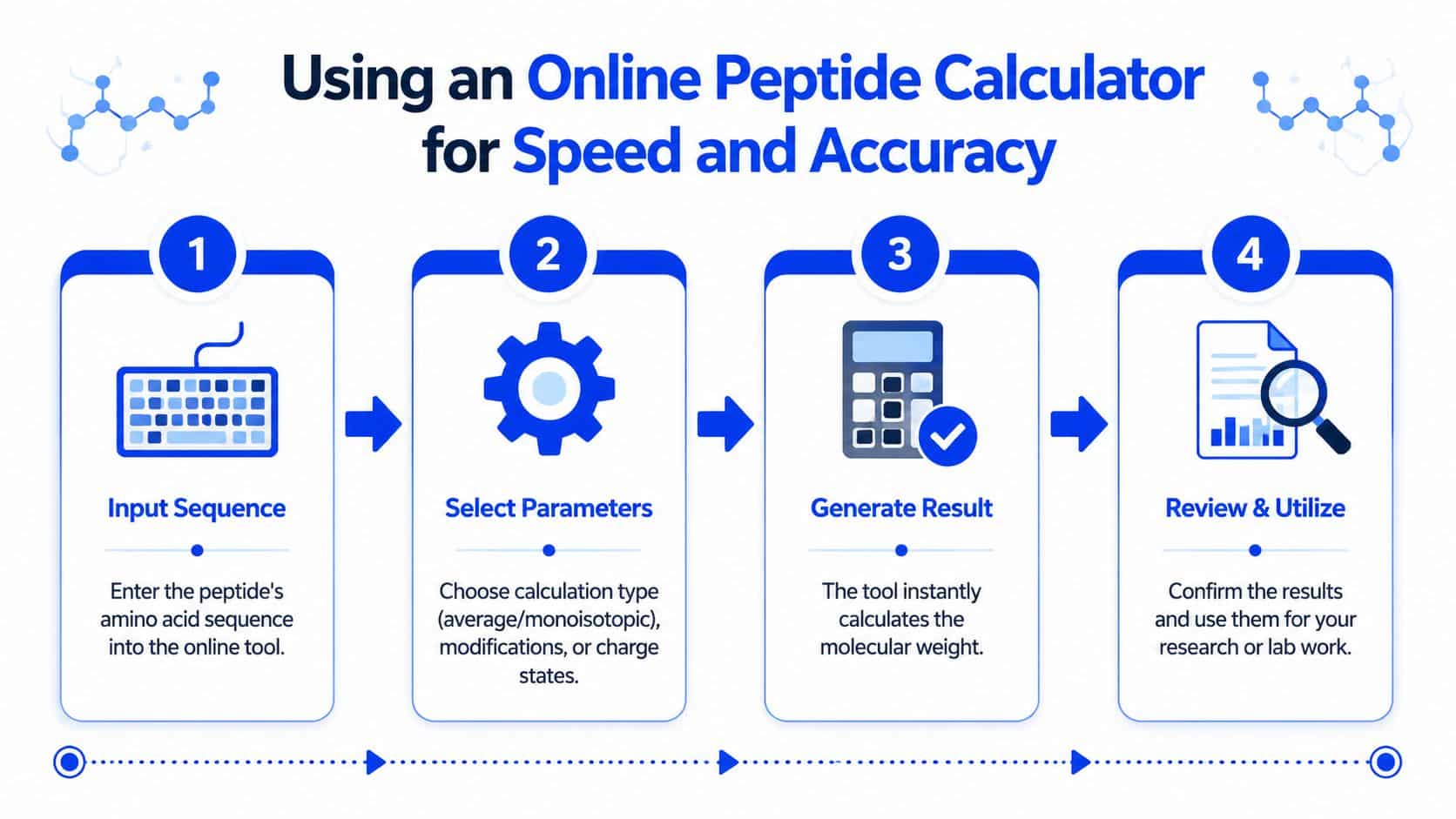
A clean workflow usually looks like this:
Enter the exact sequence
Use the correct one-letter amino acid code. Double-check residue order before doing anything else.
Set the peptide form
Confirm whether the sequence is a free peptide, acetylated peptide, amidated peptide, or another defined construct.
Add any supported modifications
Some tools explicitly support features such as phosphorylated amino acids and oxidized cysteines. Others are intended for unmodified peptides only, so assumptions matter.
Choose the mass output
Select average or monoisotopic depending on the intended use.
Review related properties
If the tool also shows pI, charge, solvent suggestions, or reconstitution-related outputs, keep those with the record. They often become useful later.
The video below shows the kind of workflow many researchers look for when using a peptide calculation tool.
Modern tools are moving beyond a single mass output. GenScript's peptide molecular weight calculator page reflects that broader workflow by pairing mass calculation with related handling outputs and interfaces researchers use during reconstitution and dosing decisions.
That's useful because the molecular weight is rarely the endpoint. In actual lab work, you usually need to turn that number into something operational.
A calculator is doing its job when it helps you answer questions like these:
What doesn't work is choosing a tool just because it returns a result quickly. Fast output is only helpful if the calculator's assumptions match the peptide in front of you.
A calculated molecular weight becomes useful only when you apply it correctly. In the lab, the number usually feeds into three decisions. You use it to prepare solutions, to interpret mass spectrometry data, and to check whether received material matches documentation.
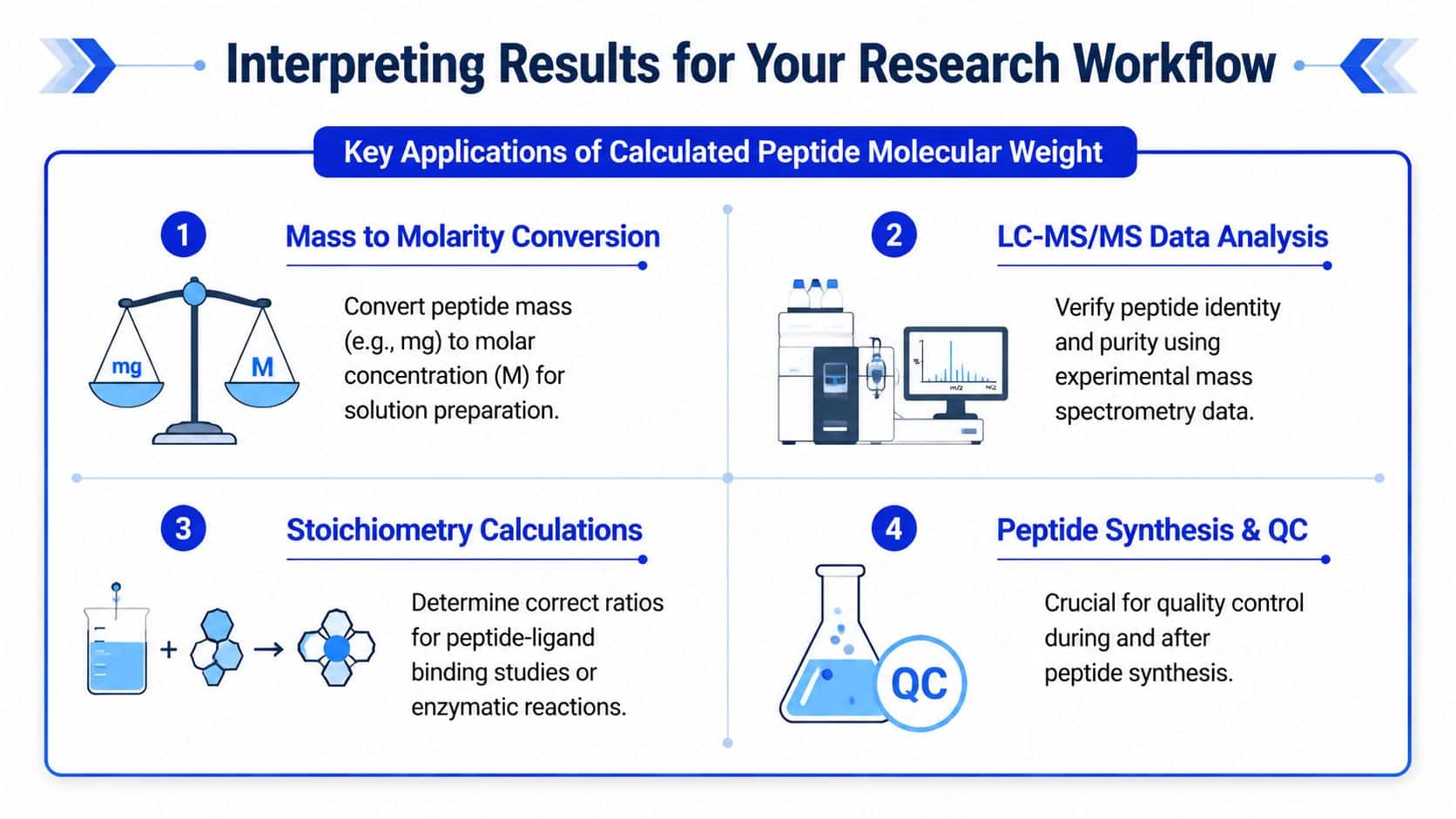
Theoretical mass meets bench reality. You don't just need a peptide's molecular weight. You need to convert the material in the vial into a usable concentration and a practical pipetting plan.
That sounds simple until the peptide behaves badly in solution. Solubility issues, aggregation, and salt form can all complicate what should have been an easy reconstitution step. GenScript's calculator page highlights this broader need by pairing mass calculation with solvent guidance and reconstitution interfaces, which reflects how researchers work.
When preparing a stock, check these before you add solvent:
What the peptide form is
Free acid, amidated, acetylated, salt-associated, and oxidized forms are not interchangeable.
What amount the vial represents
Gross powder mass and net peptide content are not always the same practical thing during reconstitution.
Whether the peptide dissolves cleanly
A theoretically correct concentration isn't useful if the peptide aggregates or remains partially undissolved.
The best stock solution on paper is worthless if the peptide hasn't actually gone into solution.
In MS work, the theoretical mass is your reference point. You compare the calculated value to the observed signal to evaluate identity and, depending on the method, support purity review or sequence confirmation.
Mass type selection is critical. If the instrument readout is being interpreted with monoisotopic assumptions, using an average molecular weight as your comparison target creates unnecessary confusion. Most “unexpected mass” conversations in peptide work are not instrument failures. They are definition failures.
A strong practice is to record the following in the same worksheet entry:
| Item to record | Why it helps |
|---|---|
| Sequence used | Confirms no residue-entry mistake occurred |
| Terminal state or modification | Prevents comparing the wrong molecular species |
| Mass type selected | Keeps average and monoisotopic results from being mixed |
| Observed analytical note | Connects the calculation to the actual experimental readout |
Supplier paperwork is often the first place researchers notice a mismatch. The COA may report a calculated mass, an observed analytical mass, purity data, and details about the peptide lot. Those records are valuable, but only if you compare like with like.
When reviewing a COA against your own calculation, ask:
This is also where practical handling separates itself from sequence math. The number from the calculator describes the peptide molecule. The material in the vial may carry additional real-world considerations related to salt forms, hydration, or handling characteristics. Good research habits account for both.
Most peptide mass errors come from a short list of preventable mistakes. They don't look dramatic when they happen, but they can waste material and confuse data review for days.
The first is ignoring terminal modifications. A peptide with a free C-terminus is not the same molecule as a C-terminal amide. The second is using the wrong mass convention for the job. Average and monoisotopic values should never be treated as interchangeable labels for the same workflow step.
Another frequent issue is sequence entry error. A single wrong residue, missing residue, or transposed position changes the result immediately. Internal structural features can also be missed, especially if the peptide includes disulfide bonding or another defined state that isn't obvious from a plain sequence line.
A calculator can only be as accurate as the sequence and chemistry you enter.
Use a short validation routine before you commit the number to an order sheet or protocol:
Read the peptide name against the sequence
Make sure labels like acetylated, amidated, phosphorylated, or oxidized appear in the calculation setup.
Check the intended application
Confirm whether the output should be monoisotopic for analytical interpretation or average for broader handling use.
Compare with supplier documentation
The sequence, stated modifications, and listed mass should align logically.
Run a second check
If the peptide is high value or the experiment is sensitive, verify with another trusted method or calculator.
That final review step is cheap insurance.
The molecular weight describes the peptide molecule itself. The weight of lyophilized material in a vial can reflect more than just the pure peptide molecule, including practical factors related to the final material form. That's why vial mass and calculated peptide molecular weight shouldn't be treated as identical ideas.
A disulfide-linked peptide is not the same mass as the fully reduced version. If your peptide contains cysteines that form a defined disulfide bond, make sure the calculation reflects that bonded state rather than assuming all cysteines remain reduced and free.
For routine solution preparation, researchers often discuss the peptide using average molecular weight. For high-resolution analytical comparison, monoisotopic mass is usually the more relevant number. The safest approach is to match the mass convention to the workflow and record which one you used.
Treat that as a warning sign. Some peptide calculators are intended for simple unmodified peptides, while others explicitly support modification-aware inputs. If the tool doesn't state its assumptions clearly, don't use the result for formulation, QC, or ordering decisions without independent verification.
Because mass alone rarely answers the practical bench question. Researchers usually need to know how the peptide will behave during reconstitution and handling, not just what the sequence weighs in theory.
If you're sourcing research peptides and want transparent documentation to support mass checks, COA review, and bench planning, explore Peptide Warehouse USA. Their catalog is built for research use, with batch-specific documentation that helps labs verify what they're working with before they move into solution prep, analytical review, or preclinical workflows. Learn more and explore options that fit your lab's peptide research needs.



